Introduction
Proteins in cells and tissues fulfill a large and diverse set of functions that enable the most basic, as well the most sophisticated processes to take place in living organisms.
Protein functions result from the inherent structural complexity of these molecules and are based, at least partially, on the capability of each protein to bind a certain ligand (or multiple specific ligands).
Such ligands might be small organic molecules (~600 Da or less), macromolecules or even elemental ions.
The roles played by the ligands are equally diverse, and include the following:
-
Catalysis. Enzymes act on substrates, turning them into products. Both substrates and products can be considered as ligands, and may be small molecules, peptides, or macromolecules.
-
Regulation. Many small organic molecules are routinely used by cells to regulate the activity of metabolic enzymes, signal transduction proteins, or other key proteins. The regulation may be simple, as in the case of product inhibition used in metabolic pathways, or more sophisticated, as in the case of hormone-activated control over key cellular processes.
-
Communication. Ligands may participate at different points along cellular communication pathways: first messenger (hormone, neurotransmitter, or local mediator), second messenger (e.g., cAMP, IP3), and downstream regulator.
-
Protein trafficking. Certain ligands serve as means by which organelles or other macromolecules are recognized by proteins.
-
Prosthetic groups. Certain ligands bind tightly to proteins and help them execute certain functions.
-
Defense and offense. Certain ligands act as toxins that attack other cells.
Protein substates
Proteins are held together by relatively weak bonds — like Hydrogen bond and van der Waals forces — rather than just hard covalent “glues.”
Think of a protein not as a rigid, static structure, but as a restless, “breathing” machine. Even when a protein is performing a specific job, it isn’t locked into one single shape.
Instead, it constantly vibrates and shifts between slightly different versions of itself:∫
-
Thermal Fluctuations: At physiological temperatures (like inside your body), atoms are constantly bumping into each other. This provides enough “kick” for the protein to overcome the small energy barriers between substates.
-
Statistical Mechanics: A protein doesn’t choose a shape; it explores all available shapes that its energy allows. At any given moment, a population of the same protein will be spread across many different substates.
Proteins shift spontaneously among multiple conformations, called ‘substates’.
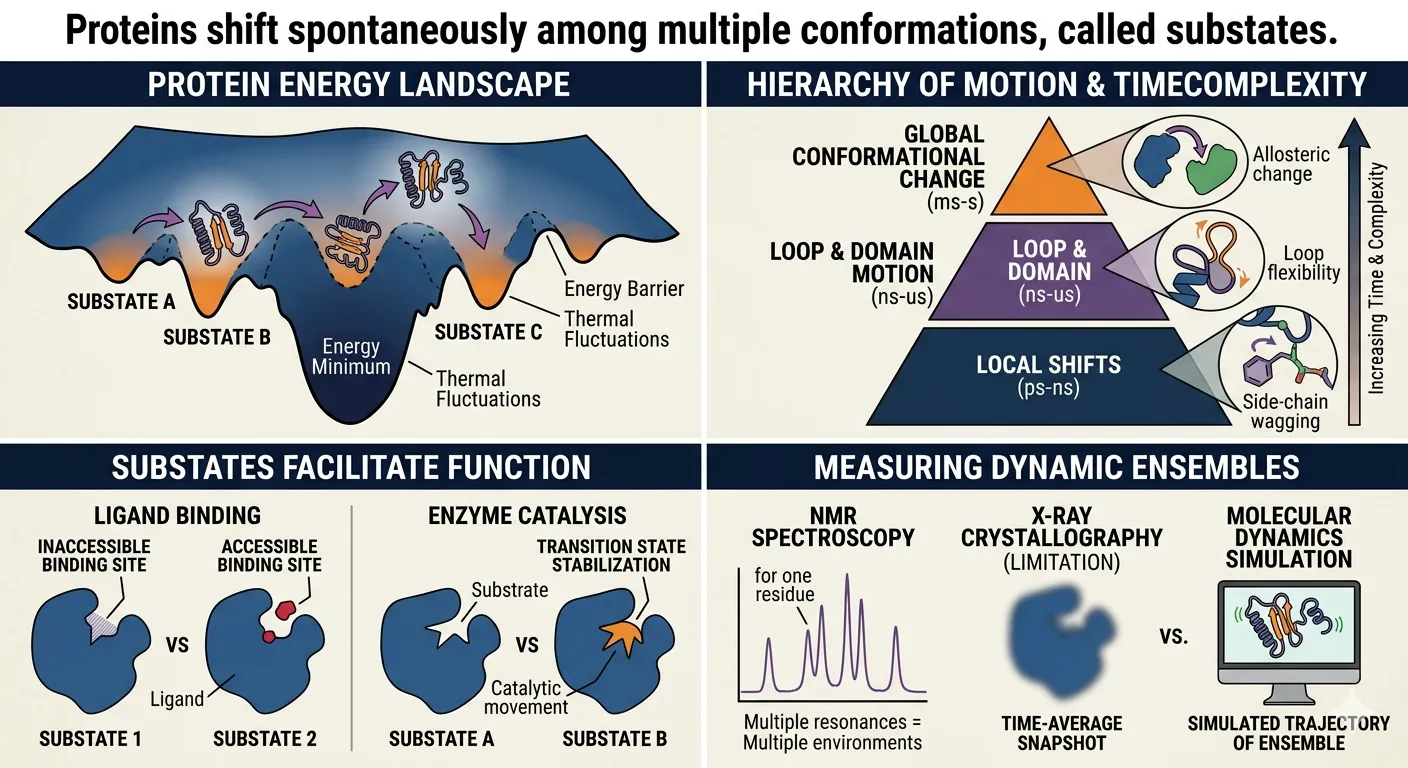
To understand substates, you have to look at the Energy Landscape.
Imagine a rugged mountain range where the “altitude” represents the energy level of the protein.
-
The Global Minimum: This is the deepest valley, representing the protein’s most stable, “native” fold.
-
Substates (The Pits): Within that big valley, there are hundreds of tiny little divots or “pits.” Each pit is a conformational substate.
-
The Shift: Because proteins exist at temperatures above absolute zero, they have thermal energy. This energy allows them to “jiggle” out of one tiny pit and fall into the one next to it.
This spontaneous shifting isn’t just “noise”; it’s vital for biological life.
| Feature | Impact on Function |
|---|---|
| Ligand Binding | A protein might shift into a substate that “opens a door” to let a drug or molecule in. |
| Enzyme Catalysis | Substates allow an enzyme to “flex” its muscles to put pressure on a chemical bond, breaking it more easily. |
| Precision | By shifting through substates, proteins can fine-tune their activity based on the pH or temperature of their environment. |
These shifts happen at different scales, which scientists usually categorize by speed:
- Local Shifts (Picoseconds): Tiny movements, like a single side-chain of an amino acid wagging back and forth.
- Medium Shifts (Nanoseconds): Small loops or “arms” of the protein moving.
- Global Shifts (Micro-to-Milliseconds): Large “domain” movements where whole sections of the protein hinge or twist. This is often where the real biological “action” happens.
If a protein were a statue, it would be useless. Because it is a collection of substates, it acts more like a dynamic ensemble, constantly “sampling” different shapes to find the one that fits the task at hand.
Conformational selection model
Traditionally, the “Induced Fit” model — where the protein changes its shape after the ligand starts to bind (like a glove stretching to fit a hand) — was the dominant theory.
However, we now know that proteins are often more proactive.
The Conformational Selection Model suggests that the protein is already “trying on” different shapes before the ligand even arrives.
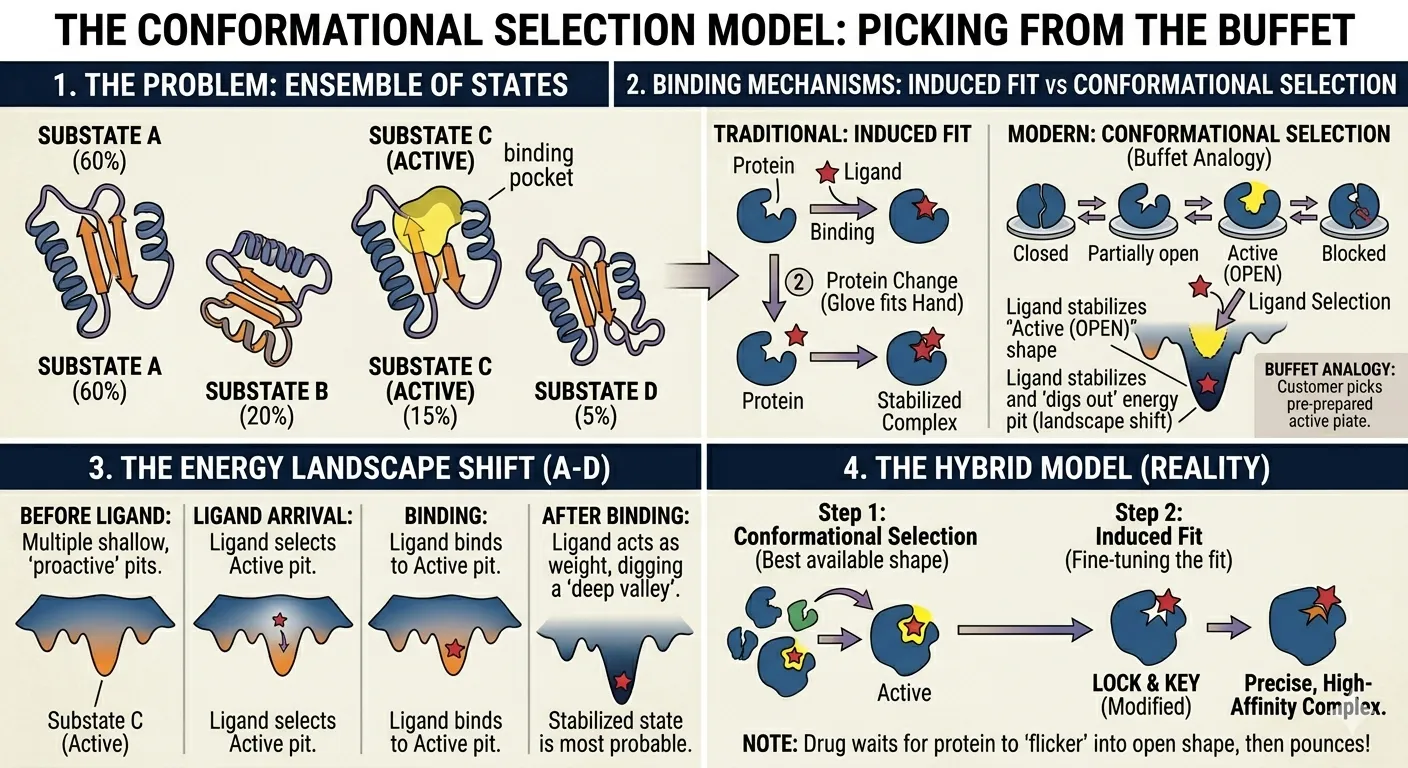
Picking from the Buffet
Think of the protein as a person at a buffet with multiple plates (substates) already laid out.
- Some plates have “closed doors” (inactive shapes).
- A few plates have “open doors” (active shapes).
When the ligand (the customer) arrives, it doesn’t wait for a new dish to be cooked. Instead, it “selects” the plate that is already open and fits it perfectly.
By binding to that specific open shape, the ligand stabilizes it. This means the protein “stays” in that active shape much longer than it would have otherwise.
The Energy Shift
In the language of the Energy Landscape, the ligand acts like a heavy weight:
- Before the ligand arrives, the active substate might be a “shallow pit” (not very stable).
- Once the ligand binds, it “digs out” that pit, making it the deepest valley in the mountain range.
- Now, the protein is much more likely to be found in this state than any other.
When selection isn’t enough: The Hybrid Model
Real life is often a mix of both models.
- Selection First: The ligand finds the “best available” shape from the protein’s ensemble.
- Induced Fit Second: Once they meet, they might both “squish” slightly to reach an even tighter, more perfect fit.
This two-step process allows proteins to be both flexible and incredibly precise.
Why does this matter? This model explains how some drugs can bind to proteins even when the “main” shape of the protein looks like it’s blocked. The drug just waits for the protein to spontaneously “flicker” into the right shape, and then it pounces!
Binding site
Evolution has equipped proteins with binding sites that match their natural ligands almost perfectly, thus optimizing the binding interactions.
Proteins bind a diverse set of ligands, and it is therefore not surprising that binding sites are equally diverse.
The binding site-ligand match is based on two main features: geometry and electrostatics.
The former has traditionally been believed to be the dominant factor in ligand compatibility. However, as a recent study has demonstrated, different protein binding sites for the same ligand exhibit greater geometric variability than can be accounted for by the conformational variability of the ligand. This suggests that geometry alone is insufficient to enable a binding site to recognize the correct ligand.
Since a given geometric or electrostatic pattern can be achieved using different binding site architectures, there is no single structural motif (i.e., loops, turns, helices, and sheets) dedicated to the binding function. That is, each of these motifs may be adapted to complement the ligand.
Geometric complementarity
The binding site is designed to geometrically complement the ligand’s three-dimensional structure.
Hence, binding sites for small molecules tend to be shaped as small and deep depressions, whereas those for peptides and proteins are larger and flatter (although in some cases the site and ligand are intertwined).
The geometrical match optimizes all noncovalent interactions that mediate the binding, particularly the short-range van der Waals interactions. The match, however, is not perfect, and as explained above, ligand-induced fit of the binding site occurs in many cases in order to optimize binding interactions.
Yet, even this does not create perfect complementarity, and cases have been recorded in which binding creates strain in the protein, ligand, and both. Such strain involves an energy cost that is often associated with deformation in one or two of the binding partners.
This may be beneficial; for example, it may help enzymes to force certain geometries on their substrates, which in turn makes it easier for the latter to turn into products. Evidently, the binding interactions are sufficient to overcompensate for the strain energy (or else the complex would not form).
Thus, the unique capability of the native (ligand-bound) structure of the protein is not necessarily its capacity to maximize favorable interactions, but rather its capacity to do so while balancing the resulting unfavorable effects, such as strain, which are required for the protein’s function.
Electrostatic complementarity
When a ligand carries an electric charge in its binding region, the corresponding protein binding site tends to carry the opposite charge.
This electrostatic match increases the binding specificity, and since electrostatic forces are long-ranged, the match may also decrease the diffusion time of the ligand to the binding site.
As in protein folding, the specificity provided to protein-ligand binding by electrostatic interactions results from two qualities:
-
The strong dependence of these interactions on the local dielectric. While a protein and its ligand may form complexes of different conformations, only the right one pairs all the right polar groups in the two binding partners. This pairing compensates for the desolvation of these groups due to the drop in dielectric upon binding.
-
The geometric dependence of hydrogen bonds, 𝜋-𝜋 interactions, and other interactions that involve molecular orbitals (e.g., n → 𝜋∗ interactions, X-bonds, and interactions involving the low-lying 𝜎∗ orbitals of the sulfur atom).